La herramienta Blast,
de donde provienen nuestras secuencias
Otra de las tareas que descubrimos el pasado lunes 17 fue entender el origen de nuestras secuencias … que no es otro que la base de datos del NCBI donde se encuentra depositado todo (o casi todo) lo secuenciado hasta la fecha… (y anotado): https://www.ncbi.nlm.nih.gov/

Interfaz de la web del NCBI y su herramienta comparativa y de búsqueda “Blast” que puede utilizar secuencias de nucleótidos y de proteínas para la búsqueda…
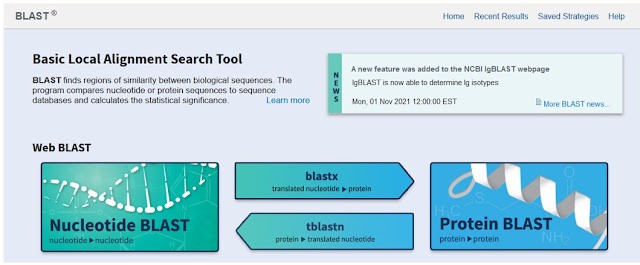


Donde analizando “grafic summary”, alignment results, “distance tree results” podemos entender lo conservado y el número de genes que hay como el nuestro (que son muchos)… . Y entre ellas, está por supuesto la primera que es nuestro gen (nuestra proteína).
Por tanto, es tb una buena herramienta de chequeo del tamaño de genes adecuado que estamos “anotando”.
Una vez identificados los genes debemos de ordenarlos de mayor a menor parecido y confirmar el paralelismo en distancia entre el gen efector y el gen RT… Algo parecido se ha realizado en el artículo del año anterior (fijaros en la figura 5 del artículo del año pasado en recursos: Nidame-Rodriguez y col 2021). Para ello hemos aprendido a utilizar dentro de la herramienta “align del clone manager cómo ordenar nuestras comparaciones (de mayor a menor parecido con nuestro locus modelo (la entrada 1352)

Nuevas tareas nos esperan: terminar de ordenar toda la comparativa del grupo A y anotar e identificar los otros 3 grupos: B, C y D.
Esta labor va a ser importante ya que nos centrará en:
la identificación de la estructura del retrón de este sistema, que como ya hemos visto
nadie ha descrito todavía!!!!
Nos vamos a cercando a nuestra meta!!




Buenas Paco, tenemos una duda respecto al alineamiento de las proteínas: ¿si las ordenamos por parecido, deberían de ser paralelos el RT y el efector de una misma secuencia? Hemos alineado y ordenado los RT y efectores del grupo B, y aunque algunos si que nos salen paralelos, la mayoría no.
ResponderEliminarPor otra parte, ¿deberíamos alinear las proteínas de cada grupo por separado o nos esperamos a hacerlo con todos los grupos a la vez?
Hola a las dos:
ResponderEliminaracabo de ver vuestros análisis, y creo que va a ser mejor el estudio por grupos por separado, sin incluir "siempre" el locus de referencia 1352. De esta manera es muy posible que las distancias entre ellos (parecidos) sean más informativas. Así, debemos de ordenar el alineamiento de las entradas A (1352-1359), B (1325-1333), C(1296-1300) y D(1336-1342). Utilizando incialmente como referencia el 1º de cada serie.