El pasado lunes tuvimos una reunión…. Cómo diría… una reunión de despropósitos…
Lo que tenía que ser una “clase” aclaratoria de conceptos, resultó debido a problemas varios (de Internet pero no sólo) “todo confusión”… Todo???
No, todo no. Descubrimos lo complejo de la labor en la que andamos embarcados… Y es que los programas de plegamiento de los ácidos nucléicos (RNA-fold) no siempre aciertan… . Y es que Paco nos explicó que el plegamiento final de un ARN depende tb de interacciones con otras proteínas, presencia de iones, conocimiento de otras estructuras parecidas, etc. Sin embargo, los “algoritmos” de plegamiento de los ordenadores se basan exclusivamente en encontrar el plegamiento “energéticamente más estable” para una determinada secuencia. Y por tanto a veces hay que corregirlo…
Pero corregirlo … cómo???
Para que entendamos a lo que me refiero debemos de adentrarnos en todas las estructuras de las secuencias del grupo D. Debemos de analizar… cual es la estructura que sale con cada una de las secuencias y compararla con la estructura que muestra cuando subimos el alineamiento (examinad vuestras notas) y enviadme los pantallazos correspondientes.
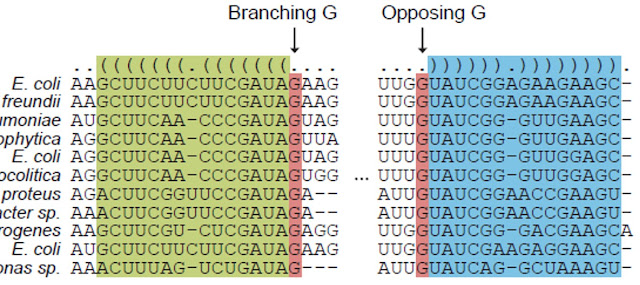
Que nos sale??? Que estructura nos da la secuencia 1341?? Que nos da cuando subimos todo el alineamiento 1342-1336 ¿?. (mirad la figura 1).

Si logramos entender lo que sucede … ya estamos muy cerca de nuestro objetivo!!!
Para ello os aconsejo que miréis la nomenclatura viena de ambas secuencias… Observareis que en el caso B es fácil encontrar las Gs de nuestro retron y menos fácil (imposible) en el caso de la 1341 aislada
Esta es la base de nuestra manera de enfrentarnos al problema de corregir los plegamientos. Y entender lo que pretendemos en nuestro trabajo.
Para corregirlo es super importante que entendamos la “nomenclatura Viena”. Que no es otra que una nomenclatura que predice las interacciones de bases en una molécula de RNA a base de “.” : no interacción y paréntesis”( )” : interacción.

Figura 2: Nomenclatura Viena ejemplo.
Y usar un programa (gratuito) para visualizar de estructuras. El programa es VARNAv3-93.jar nos lo podemos descargar desde: http://varna.lri.fr/index.php?lang=en&page=downloads&css=varna

Este programa nos permite proponer una determinada interacción, aunque ésta no haya sido predicha por los algoritmos de RNAfold.

Figura 4: Visualización de estructuras con el programa VARNAv3-93
El próximo día entenderemos como manejarla… . Aunque a lo mejor ya podeis ir haciendo probatinas… .
Aunque no lo parezca estamos ya muy cerca de alcanzar nuestro objetivo…
Jo!!!! Que dura es la ciencia!!!